MOE 2008.10
QuaSAR-Descriptor
Introduction
Default MOE descriptors were
calculated based on single low energy conformers of the compounds. A detailed
description of the interpretaton is given below:
2D Molecular Descriptors
2D molecular descriptors are
defined to be numerical properties that can be calculated from the connection
table representation of a molecule (e.g., elements, formal charges and
bonds, but not atomic coordinates). 2D descriptors are, therefore, not
dependent on the conformation of a molecule and are most suitable for large
database studies.
Notation and Terminology
Many descriptors make use of several fundamental quantities that can be
computed from a chemical structure. This section will define these fundamental
quantities. For purposes of illustration, the following chemical structure will
be used:
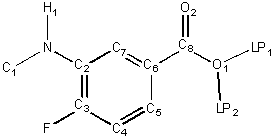
The fundamental quantities of a chemical structure depend solely on the
structure as drawn, i.e., no modifications to the structure are
implied with the exception of the addition or subtraction of hydrogen atoms to
full valence.
Z denotes the atomic number of an atom; lone
pair pseudo-atoms (LP) are given an atomic number of 0. Heavy atoms are
atoms that have an atomic number strictly greater than 1 (not
H nor LP). A trivial atom is an LP pseudo-atom or a hydrogen with exactly one heavy neighbor. In the reference
structure, H1, LP1 and LP2 are trivial.
The hydrogen count, h, of an atom is the number of hydrogens to which it is (or should be) attached. This
count includes all hydrogen atoms that are necessary to fill valence. In the
reference structure, F has h = 0, N has h = 1
and O1 has h = 1.
The heavy degree, d, of an atom is the number of heavy atoms to
which it is bonded. That is, d is the number of bonded neighbors of the
atom in the hydrogen suppressed graph. In the reference structure, F has d = 1,
C6 has d = 3 and N has d = 2.
Physical Properties
The following physical properties can be calculated from the connection table
(with no dependence on conformation) of a molecule:
|
Code |
Description |
|
apol |
Sum of the atomic polarizabilities
(including implicit hydrogens) with polarizabilities taken from [CRC 1994]. |
|
bpol |
Sum of the absolute value of the difference
between atomic polarizabilities of all bonded atoms
in the molecule (including implicit hydrogens) with
polarizabilities taken from [CRC 1994]. |
|
density |
Molecular mass density: Weight divided by vdw_vol
(amu/Å3). |
|
FCharge |
Total charge of the molecule (sum of formal
charges). |
|
mr |
Molecular refractivity (including implicit hydrogens). This property is calculated from an 11
descriptor linear model [MREF 1998] with r2 = 0.997,
RMSE = 0.168 on 1,947 small molecules. |
|
SMR |
Molecular refractivity (including implicit hydrogens). This property is an atomic contribution model
[Crippen 1999] that assumes the correct protonation state (washed structures). The model was
trained on ~7000 structures and results may vary from the mr
descriptor. |
|
Weight |
Molecular weight (including implicit hydrogens) in atomic mass units with atomic weights taken
from [CRC 1994]. |
|
logP(o/w) |
Log of the octanol/water
partition coefficient (including implicit hydrogens).
This property is calculated from a linear atom type model [LOGP 1998]
with r2 = 0.931, RMSE=0.393 on 1,827 molecules. |
|
logS |
Log of the aqueous solubility (mol/L). This
property is calculated from an atom contribution linear atom type model [Hou 2004] with r2 = 0.90,
~1,200 molecules. |
|
reactive |
Indicator of the presence of reactive groups.
A non-zero value indicates that the molecule contains a reactive group. The
table of reactive groups is based on the Oprea set
[Oprea 2000] and includes metals, phospho-, N/O/S-N/O/S single bonds, thiols,
acyl halides, Michael Acceptors, azides, esters, etc. |
|
SlogP |
Log of the octanol/water
partition coefficient (including implicit hydrogens).
This property is an atomic contribution model [Crippen 1999]
that calculates logP from the given structure;
i.e., the correct protonation state (washed
structures). Results may vary from the logP(o/w) descriptor. The
training set for SlogP was ~7000 structures. |
|
TPSA |
Polar surface area (Å2) calculated using group contributions to approximate the polar surface area from connection table information only. The parameterization is that of Ertl et al. [Ertl 2000]. |
|
vdw_vol |
van
der Waals volume (Å3)
calculated using a connection table approximation. |
|
vdw_area |
Area of van der Waals surface (Å2) calculated using a
connection table approximation. |
Subdivided
Surface Areas
The Subdivided Surface Areas are
descriptors based on an approximate accessible van der
Waals surface area (in Å2) calculation for
each atom, vi along with some other
atomic property, pi. The vi
are calculated using a connection table approximation. Each descriptor in a
series is defined to be the sum of the vi over all atoms i such that pi is in a specified
range (a,b).
In the descriptions to follow, Li
denotes the contribution to logP(o/w) for atom i as
calculated in the SlogP descriptor [Crippen 1999].
Ri denotes
the contribution to Molar Refractivity for atom i
as calculated in the SMR descriptor [Crippen 1999]. The ranges were
determined by percentile subdivision over a large collection of compounds.
|
Code |
Description |
|
SlogP_VSA0 |
Sum of vi
such that Li <= -0.4. |
|
SlogP_VSA1 |
Sum of vi
such that Li is in (-0.4,-0.2]. |
|
SlogP_VSA2 |
Sum of vi
such that Li is in (-0.2,0]. |
|
SlogP_VSA3 |
Sum of vi
such that Li is in (0,0.1]. |
|
SlogP_VSA4 |
Sum of vi
such that Li is in (0.1,0.15]. |
|
SlogP_VSA5 |
Sum of vi
such that Li is in (0.15,0.20]. |
|
SlogP_VSA6 |
Sum of vi
such that Li is in (0.20,0.25]. |
|
SlogP_VSA7 |
Sum of vi
such that Li is in (0.25,0.30]. |
|
SlogP_VSA8 |
Sum of vi
such that Li is in (0.30,0.40]. |
|
SlogP_VSA9 |
Sum of vi
such that Li > 0.40. |
|
SMR_VSA0 |
Sum of vi
such that Ri is in [0,0.11]. |
|
SMR_VSA1 |
Sum of vi
such that Ri is in (0.11,0.26]. |
|
SMR_VSA2 |
Sum of vi
such that Ri is in (0.26,0.35]. |
|
SMR_VSA3 |
Sum of vi
such that Ri is in (0.35,0.39]. |
|
SMR_VSA4 |
Sum of vi
such that Ri is in (0.39,0.44]. |
|
SMR_VSA5 |
Sum of vi
such that Ri is in (0.44,0.485]. |
|
SMR_VSA6 |
Sum of vi
such that Ri is in (0.485,0.56]. |
|
SMR_VSA7 |
Sum of vi
such that Ri > 0.56. |
Atom Counts
and Bond Counts
The atom count and bond count
descriptors are functions of the counts of atoms and bonds (subdivided
according to various criteria).
|
Code |
Description |
|
a_aro |
Number of aromatic atoms. |
|
a_count |
Number of
atoms (including implicit hydrogens). This is
calculated as the sum of (1 + hi) over all
non-trivial atoms i. |
|
a_heavy |
Number of
heavy atoms #{Zi | Zi > 1}. |
|
a_ICM |
Atom
information content (mean). This is the entropy of the element distribution in
the molecule (including implicit hydrogens but not
lone pair pseudo-atoms). Let ni
be the number of occurrences of atomic number i
in the molecule. Let pi = ni / n
where n is the sum of the ni.
The value of a_ICM is the negative of the sum over all i of pi log pi. |
|
a_IC |
Atom
information content (total). This is calculated to be a_ICM times n. |
|
a_nH |
Number of
hydrogen atoms (including implicit hydrogens). This
is calculated as the sum of hi over all non-trivial atoms i plus the number of non-trivial hydrogen atoms. |
|
a_nB |
Number of
boron atoms: #{Zi | Zi = 5}. |
|
a_nC |
Number of
carbon atoms: #{Zi | Zi = 6}. |
|
a_nN |
Number of
nitrogen atoms: #{Zi | Zi = 7}. |
|
a_nO |
Number of
oxygen atoms: #{Zi | Zi = 8}. |
|
a_nF |
Number of
fluorine atoms: #{Zi | Zi = 9}. |
|
a_nP |
Number of
phosphorus atoms: #{Zi | Zi = 15}. |
|
a_nS |
Number of
sulfur atoms: #{Zi | Zi = 16}. |
|
a_nCl |
Number of
chlorine atoms: #{Zi | Zi = 17}. |
|
a_nBr |
Number of
bromine atoms: #{Zi | Zi = 35}. |
|
a_nI |
Number of
iodine atoms: #{Zi | Zi = 53}. |
|
b_1rotN |
Number of
rotatable single bonds. Conjugated single bonds are
not included (e.g., ester and peptide bonds). |
|
b_1rotR |
Fraction
of rotatable single bonds: b_1rotN divided by b_heavy. |
|
b_ar |
Number of aromatic bonds. |
|
b_count |
Number of
bonds (including implicit hydrogens). This is
calculated as the sum of (di/2 + hi)
over all non-trivial atoms i. |
|
b_double |
Number of
double bonds. Aromatic bonds are not considered to be double bonds. |
|
b_heavy |
Number of
bonds between heavy atoms. |
|
b_rotN |
Number of
rotatable bonds. A bond is rotatable
if it has order 1, is not in a ring, and has at least two heavy neighbors. |
|
b_rotR |
Fraction
of rotatable bonds: b_rotN divided by b_heavy. |
|
b_single |
Number of
single bonds (including implicit hydrogens).
Aromatic bonds are not considered to be single bonds. |
|
b_triple |
Number of
triple bonds. Aromatic bonds are not considered to be triple bonds. |
|
chiral |
The
number of chiral centers.
|
|
chiral_u |
The
number of unconstrained chiral centers.
|
|
lip_acc |
The
number of O and N atoms. |
|
lip_don |
The
number of OH and NH atoms. |
|
lip_druglike |
One if and
only if lip_violation < 2 otherwise zero. |
|
lip_violation |
The
number of violations of Lipinski's Rule of Five [Lipinski 1997]. |
|
nmol |
The
number of molecules (connected components). |
|
opr_brigid |
The
number of rigid bonds from [Oprea 2000]. |
|
opr_leadlike |
One if
and only if opr_violation < 2 otherwise zero. |
|
opr_nring |
The
number of ring bonds from [Oprea 2000]. |
|
opr_nrot |
The
number of rotatable bonds from [Oprea 2000].
|
|
opr_violation |
The number
of violations of Oprea's lead-like test [Oprea 2000]. |
|
rings |
The number of rings. |
|
VAdjMa |
Vertex adjacency information (magnitude): 1 + log2 m where m is the number of heavy-heavy bonds. If m is zero, then zero is returned. |
|
VAdjEq |
Vertex
adjacency information (equality): -(1-f)log2(1-f) - f log2 f
where f = (n2 - m) / n2,
n is the number of heavy atoms and m is the number of
heavy-heavy bonds. If f is not in the open interval (0,1), then 0 is returned. |
Kier&Hall
Connectivity and Kappa Shape Indices
For a heavy atom i let vi = (pi - hi) / (Zi - pi - 1) where
pi is the number of s and p valence electrons of atom i. The Kier and Hall chi connectivity indices are
calculated from the heavy atom degree di
(number of heavy neighbors) and vi.
The Kier and Hall kappa molecular shape indices [Hall 1991] compare the
molecular graph with minimal and maximal molecular graphs, and are intended to
capture different aspects of molecular shape. In the following description, n
denotes the number of atoms in the hydrogen suppressed graph, m is the
number of bonds in the hydrogen suppressed graph and a is the sum of (ri/rc - 1) where ri is the
covalent radius of atom i, and rc is the covalent radius of a carbon
atom. Also, let p2 denote the number of paths of length 2
and p3 the number of paths of length 3.
|
Code |
Description |
|
chi0 |
Atomic
connectivity index (order 0) from [Hall 1991] and [Hall 1977]. This
is calculated as the sum of 1/sqrt(di) over all heavy atoms i with di > 0. |
|
chi0_C |
Carbon
connectivity index (order 0). This is calculated as the sum of 1/sqrt(di) over
all carbon atoms i with di > 0. |
|
chi1 |
Atomic connectivity
index (order 1) from [Hall 1991] and [Hall 1977]. This is
calculated as the sum of 1/sqrt(didj)
over all bonds between heavy atoms i and j
where i < j. |
|
chi1_C |
Carbon
connectivity index (order 1). This is calculated as the sum of 1/sqrt(didj) over all bonds between
carbon atoms i and j where i < j. |
|
chi0v |
Atomic
valence connectivity index (order 0) from [Hall 1991] and
[Hall 1977]. This is calculated as the sum of 1/sqrt(vi)
over all heavy atoms i with vi > 0. |
|
chi0v_C |
Carbon valence
connectivity index (order 0). This is calculated as the sum of 1/sqrt(vi) over all carbon atoms i with vi > 0. |
|
chi1v |
Atomic
valence connectivity index (order 1) from [Hall 1991] and
[Hall 1977]. This is calculated as the sum of 1/sqrt(vivj)
over all bonds between heavy atoms i and j
where i < j. |
|
chi1v_C |
Carbon
valence connectivity index (order 1). This is calculated as the sum of
1/sqrt(vivj) over all
bonds between carbon atoms i and j
where i < j. |
|
Kier1 |
First
kappa shape index: (n-1)2 / m2
[Hall 1991]. |
|
Kier2 |
Second
kappa shape index: (n-1)2 / m2
[Hall 1991]. |
|
Kier3 |
Third
kappa shape index: (n-1) (n-3)2 / p32
for odd n, and (n-3) (n-2)2 / p32
for even n [Hall 1991]. |
|
KierA1 |
First
alpha modified shape index: s (s-1)2 / m2
where s = n + a [Hall 1991]. |
|
KierA2 |
Second
alpha modified shape index: s (s-1)2 / m2
where s = n + a [Hall 1991]. |
|
KierA3 |
Third
alpha modified shape index: (n-1) (n-3)2 /
p32 for odd n, and (n-3) (n-2)2 / p32
for even n where s = n + a
[Hall 1991]. |
|
KierFlex |
Kier molecular flexibility index: (KierA1) (KierA2) / n [Hall 1991]. |
|
zagreb |
|
Adjacency and
Distance Matrix Descriptors
The adjacency matrix, M,
of a chemical structure is defined by the elements [Mij]
where Mij is 1 if atoms i and j are bonded and zero otherwise. The distance
matrix, D, of a chemical structure is defined by the elements [Dij] where Dij
is the length of the shortest path from atoms i
to j; zero is used if atoms i and j
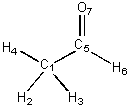
are not part of the same connected component. The adjacency matrix of CH3CH=O
is displayed on the left and its distance matrix is displayed on the right
(below):
C1 0 1 1 1 1 0 0 0 1 1 1 1 2 2 H2 1 0 0 0 0 0 0 1 0 2 2 2 3 3 H3 1 0 0 0 0 0 0 1 2 0 2 2 3 3 H4 1 0 0 0 0 0 0 1 2 2 0 2 3 3 C5 1 0 0 0 0 1 1 1 2 2 2 0 1 1 H6 0 0 0 0 1 0 0 2 3 3 3 1 0 2 O7 0 0 0 0 1 0 0 2 3 3 3 1 2 0 |
|
Petitjean [Petitjean
1992] defines the eccentricity of a vertex to be the longest path from
that vertex to any other vertex in the graph. The graph radius is the
smallest vertex eccentricity in the graph and the graph diameter as the
largest vertex eccentricity. These values are calculated using the distance
matrix and are used for several descriptors described below.
The following descriptors are
calculated from the distance and adjacency matrices of the heavy atoms:
|
Code |
Description |
|
balabanJ |
Balaban's
connectivity topological index [Balaban 1982].
|
|
BCUT_PEOE_0 |
The BCUT descriptors
[Pearlman 1998] are calculated from the eigenvalues
of a modified adjacency matrix. Each ij
entry of the adjacency matrix takes the value 1/sqrt(bij)
where bij is the formal bond
order between bonded atoms i and j.
The diagonal takes the value of the PEOE partial charges. The resulting eigenvalues are sorted and the smallest, 1/3-ile, 2/3-ile
and largest eigenvalues are reported. |
|
BCUT_SLOGP_0 |
The BCUT
descriptors using atomic contribution to logP
(using the Wildman and Crippen SlogP
method) instead of partial charge. |
|
BCUT_SMR_0 |
The BCUT
descriptors using atomic contribution to molar refractivity (using the Wildman
and Crippen SMR method) instead of partial charge. |
|
diameter |
Largest
value in the distance matrix [Petitjean 1992].
|
|
petitjean |
Value of
(diameter - radius) / diameter. |
|
GCUT_PEOE_0 |
The GCUT descriptors
are calculated from the eigenvalues of a modified
graph distance adjacency matrix. Each ij
entry of the adjacency matrix takes the value 1/sqr(dij)
where dij is the (modified) graph
distance between atoms i and j. The
diagonal takes the value of the PEOE partial charges. The resulting eigenvalues are sorted and the smallest, 1/3-ile, 2/3-ile
and largest eigenvalues are reported. |
|
GCUT_SLOGP_0 |
The GCUT descriptors
using atomic contribution to logP (using the
Wildman and Crippen SlogP
method) instead of partial charge. |
|
GCUT_SMR_0 |
The GCUT
descriptors using atomic contribution to molar refractivity (using the
Wildman and Crippen SMR method) instead of partial
charge. |
|
petitjeanSC |
Petitjean
graph Shape Coefficient as defined in [Petitjean 1992]:
(diameter - radius) / radius. |
|
radius |
If ri is the largest matrix entry in row
i of the distance matrix D, then
the radius is defined as the smallest of the ri
[Petitjean 1992]. |
|
VDistEq |
If m
is the sum of the distance matrix entries then VdistEq is defined to be the sum of log2 m - pi log2 pi / m
where pi is the number of distance matrix entries equal to i. |
|
VDistMa |
If m
is the sum of the distance matrix entries then VDistMa is defined to be the sum of log2 m - Dij log2 Dij / m over all i and j. |
|
wienerPath |
Wiener
path number: half the sum of all the distance matrix entries as defined in [Balaban 1979] and [Wiener 1947]. |
|
wienerPol |
Wiener
polarity number: half the sum of all the distance matrix entries with a value
of 3 as defined in [Balaban 1979]. |
Pharmacophore
Feature Descriptors
The Pharmacophore
Atom Type descriptors consider only the heavy atoms of a molecule and assign a
type to each atom. That is, hydrogens are suppressed
during the calculation. The atom typing mechanism is located in the file $MOE/lib/svl/ph4.svl/ph4type.svl which is a rule-based system for
assigning pharmacophore features to atoms. The
feature set is Donor, Acceptor, Polar (both Donor and Acceptor), Positive
(base), Negative (acid), Hydrophobe and Other.
Assignments may take into account implied protonation,
deprotonation, keto/enol
considerations and tautomerism at a biologically
relevant pH. For example, -COOH will be typed in its deprotonated
form regardless of how the structure is stored.
|
Code |
Description |
|
a_acc |
Number of
hydrogen bond acceptor atoms (not counting acidic atoms but counting atoms
that are both hydrogen bond donors and acceptors such as -OH). |
|
a_acid |
Number of acidic atoms. |
|
a_base |
Number of basic atoms. |
|
a_don |
Number of
hydrogen bond donor atoms (not counting basic atoms but counting atoms that
are both hydrogen bond donors and acceptors such as -OH). |
|
a_hyd |
Number of hydrophobic atoms. |
|
vsa_acc |
Approximation
to the sum of VDW surface areas (Å2) of pure hydrogen bond
acceptors (not counting acidic atoms and atoms that are both hydrogen bond
donors and acceptors such as -OH). |
|
vsa_acid |
Approximation
to the sum of VDW surface areas of acidic atoms (Å2). |
|
vsa_base |
Approximation
to the sum of VDW surface areas of basic atoms (Å2). |
|
vsa_don |
Approximation
to the sum of VDW surface areas of pure hydrogen bond donors (not counting
basic atoms and atoms that are both hydrogen bond donors and acceptors such
as -OH) (Å2). |
|
vsa_hyd |
Approximation
to the sum of VDW surface areas of hydrophobic atoms (Å2). |
|
vsa_other |
Approximation
to the sum of VDW surface areas (Å2) of atoms typed as
"other". |
|
vsa_pol |
Approximation
to the sum of VDW surface areas (Å2) of polar atoms (atoms that
are both hydrogen bond donors and acceptors), such as -OH. |
Partial
Charge Descriptors
Descriptors that depend on the
partial charge of each atom of a chemical structure require calculation of those
partial charges. An unfortunate complication is the fact that there are
numerous methods of calculating partial charges. Rather than enforce a
particular method, MOE provides several versions of most of the
charge-dependent descriptors. The only difference between these variants is the
source of the partial charges. The following variants are supported: PEOE, Q
(described below).
PEOE. The Partial Equalization of Orbital Electronegativities (PEOE) method of calculating atomic
partial charges [Gasteiger 1980] is a method in
which charge is transferred between bonded atoms until equilibrium. To
guarantee convergence, the amount of charge transferred at each iteration is
damped with an exponentially decreasing scale factor. The amount of charge transferred,
dqij, between atoms i and j when Xi > Xj is
dqij = (1/2k) (Xi
- Xj) / Xj+
where Xj+
is the electronegativity of the positive ion of atom j;
Xi is the electronegativity of atom
i (quadratically
dependent on partial charge); and k is the iteration number of the
algorithm. Electronegativity values are determined by
parameterization found in the SVL source code file $MOE/lib/svl/calc.svl/charge.svl. The PEOE charges depend only on the
connectivity of the input structures: elements, formal charges and bond orders.
Descriptors using the PEOE charges are prefixed with PEOE_.
Q. Descriptors prefixed with Q_ use the partial charges stored with each
structure in the database. In other words, no partial charge calculation is
made and it is assumed that some external program has been used to calculate
the atomic partial charges. This dependence can be a subtle source of error if,
for example, the wrong charges are stored when descriptors are recalculated (e.g.,
when evaluating QSAR models on novel structures).
Let qi
denote the partial charge of atom i as defined
above. Let vi be the van der Waals surface area (Å2) of atom i (as calculated by a connection table
approximation). The following descriptors are calculated:
|
Code |
Description |
|
Q_PC+ |
Total
positive partial charge: the sum of the positive qi.
Q_PC+ is identical to PC+ which has been retained for
compatibility. |
|
Q_PC- |
Total
negative partial charge: the sum of the negative qi.
Q_PC- is identical to PC- which has been retained for
compatibility. |
|
Q_RPC+ |
Relative positive
partial charge: the largest positive qi
divided by the sum of the positive qi.
Q_RPC+ is identical to RPC+ which has been retained for
compatibility. |
|
Q_PRC- |
Relative
negative partial charge: the smallest negative qi
divided by the sum of the negative qi.
Q_RPC- is identical to RPC- which has been retained for
compatibility. |
|
Q_VSA_POS |
Total positive van der Waals surface area. This is the sum of the vi such that qi is non-negative. The vi are calculated using a connection table approximation. |
|
Q_VSA_NEG |
Total negative van der Waals surface area. This is the sum of the vi such that qi is negative. The vi are calculated using a connection table approximation. |
|
Q_VSA_PPOS |
Total positive polar van der Waals surface area. This is the sum of the vi such that qi is greater than 0.2. The vi are calculated using a connection table approximation. |
|
Q_VSA_PNEG |
Total negative polar van der Waals surface area. This is the sum of the vi such that qi is less than -0.2. The vi are calculated using a connection table approximation. |
|
Q_VSA_HYD |
Total hydrophobic van der Waals surface area. This is the sum of the vi such that |qi| is less than or equal to 0.2. The vi are calculated using a connection table approximation. |
|
Q_VSA_POL |
Total polar van der Waals surface area. This is the sum of the vi such that |qi| is greater than 0.2. The vi are calculated using a connection table approximation. |
|
Q_VSA_FPOS |
Fractional positive van der Waals surface area. This is the sum of the vi such that qi is non-negative divided by the total surface area. The vi are calculated using a connection table approximation. |
|
Q_VSA_FNEG |
Fractional negative van der Waals surface area. This is the sum of the vi such that qi is negative divided by the total surface area. The vi are calculated using a connection table approximation. |
|
Q_VSA_FPPOS |
Fractional positive polar van der Waals surface area. This is the sum of the vi such that qi is greater than 0.2 divided by the total surface area. The vi are calculated using a connection table approximation. |
|
Q_VSA_FPNEG |
Fractional negative polar van der Waals surface area. This is the sum of the vi such that qi is less than -0.2 divided by the total surface area. The vi are calculated using a connection table approximation. |
|
Q_VSA_FHYD |
Fractional hydrophobic van der Waals surface area. This is the sum of the vi such that |qi| is less than or equal to 0.2 divided by the total surface area. The vi are calculated using a connection table approximation. |
|
Q_VSA_FPOL |
Fractional polar van der Waals surface area. This is the sum of the vi such that |qi| is greater than 0.2 divided by the total surface area. The vi are calculated using a connection table approximation. |
|
PEOE_VSA+6 |
Sum of vi
where qi is greater than 0.3. |
|
PEOE_VSA+5 |
Sum of vi
where qi is in the range
[0.25,0.30). |
|
PEOE_VSA+4 |
Sum of vi
where qi is in the range
[0.20,0.25). |
|
PEOE_VSA+3 |
Sum of vi
where qi is in the range
[0.15,0.20). |
|
PEOE_VSA+2 |
Sum of vi
where qi is in the range
[0.10,0.15). |
|
PEOE_VSA+1 |
Sum of vi
where qi is in the range
[0.05,0.10). |
|
PEOE_VSA+0 |
Sum of vi
where qi is in the range
[0.00,0.05). |
|
PEOE_VSA-0 |
Sum of vi
where qi is in the range
[-0.05,0.00). |
|
PEOE_VSA-1 |
Sum of vi
where qi is in the range
[-0.10,-0.05). |
|
PEOE_VSA-2 |
Sum of vi
where qi is in the range
[-0.15,-0.10). |
|
PEOE_VSA-3 |
Sum of vi
where qi is in the range
[-0.20,-0.15). |
|
PEOE_VSA-4 |
Sum of vi
where qi is in the range
[-0.25,-0.20). |
|
PEOE_VSA-5 |
Sum of vi
where qi is in the range
[-0.30,-0.25). |
|
PEOE_VSA-6 |
Sum of vi
where qi is less than -0.30. |
3D Molecular
Descriptors
There are two types of 3D
molecular descriptors: those that depend on internal coordinates only and those
that depend on absolute orientation. 3D molecular descriptors are classified as
"i3D" for internal coordinate dependent 3D and "x3D" for
external coordinate dependent. A good example is the dipole moment: the
magnitude of the dipole moment does not depend on absolute orientation in
space; however, the x component of the dipole moment does depend on
absolute orientation.
Note: All the 3D descriptors operate on structures
found in the database as is; that is, no hydrogens
are added or removed. Furthermore, most descriptors assume that partial charges
are stored with the structures in the database.
MOPAC Descriptors
The MOPAC [MOPAC] descriptors are calculated by the version of MOPAC6 distributed
with MOE.
|
Code |
Description |
|
AM1_dipole |
The dipole moment calculated using the AM1
Hamiltonian [MOPAC]. |
|
AM1_E |
The total energy (kcal/mol) calculated using
the AM1 Hamiltonian [MOPAC]. |
|
AM1_Eele |
The electronic energy (kcal/mol) calculated
using the AM1 Hamiltonian [MOPAC]. |
|
AM1_HF |
The heat of formation (kcal/mol) calculated
using the AM1 Hamiltonian [MOPAC]. |
|
AM1_IP |
The ionization potential (kcal/mol)
calculated using the AM1 Hamiltonian [MOPAC]. |
|
AM1_LUMO |
The energy (eV) of
the Lowest Unoccupied Molecular Orbital calculated using the AM1 Hamiltonian
[MOPAC]. |
|
AM1_HOMO |
The energy (eV) of
the Highest Occupied Molecular Orbital calculated using the AM1 Hamiltonian
[MOPAC]. |
|
MNDO_dipole |
The dipole moment calculated using the MNDO
Hamiltonian [MOPAC]. |
|
MNDO_E |
The total energy (kcal/mol) calculated using
the MNDO Hamiltonian [MOPAC]. |
|
MNDO_Eele |
The electronic energy (kcal/mol) calculated
using the MNDO Hamiltonian [MOPAC]. |
|
MNDO_HF |
The heat of formation (kcal/mol) calculated using
the MNDO Hamiltonian [MOPAC]. |
|
MNDO_IP |
The ionization potential (kcal/mol)
calculated using the MNDO Hamiltonian [MOPAC]. |
|
MNDO_LUMO |
The energy (eV) of
the Lowest Unoccupied Molecular Orbital calculated using the MNDO Hamiltonian
[MOPAC]. |
|
MNDO_HOMO |
The energy (eV) of
the Highest Occupied Molecular Orbital calculated using the MNDO Hamiltonian
[MOPAC]. |
|
PM3_dipole |
The dipole moment calculated using the PM3
Hamiltonian [MOPAC]. |
|
PM3_E |
The total energy (kcal/mol) calculated using
the PM3 Hamiltonian [MOPAC]. |
|
PM3_Eele |
The electronic energy (kcal/mol) calculated
using the PM3 Hamiltonian [MOPAC]. |
|
PM3_HF |
The heat of formation (kcal/mol) calculated using
the PM3 Hamiltonian [MOPAC]. |
|
PM3_IP |
The ionization potential (kcal/mol)
calculated using the PM3 Hamiltonian [MOPAC]. |
|
PM3_LUMO |
The energy (eV) of
the Lowest Unoccupied Molecular Orbital calculated using the PM3 Hamiltonian
[MOPAC]. |
|
PM3_HOMO |
The energy (eV) of
the Highest Occupied Molecular Orbital calculated using the PM3 Hamiltonian
[MOPAC]. |
Surface Area,
Volume and Shape Descriptors
The following descriptors depend on the structure connectivity and
conformation (dimensions are measured in Å). The vsurf_ descriptors are similar to the VolSurf descriptors [Cruciani 2000];
these descriptors have been shown to be useful in pharmacokinetic property
prediction.
|
Code |
Description |
|
ASA |
Water accessible surface area calculated
using a radius of 1.4 A for the water molecule. A polyhedral representation
is used for each atom in calculating the surface area. |
|
dens |
Mass density: molecular weight divided by van
der Waals volume as
calculated in the vol descriptor. |
|
glob |
Globularity, or inverse condition number
(smallest eigenvalue divided by the largest eigenvalue) of the covariance matrix of atomic
coordinates. A value of 1 indicates a perfect sphere while a value of 0
indicates a two- or one-dimensional object. |
|
pmi |
Principal moment of inertia. |
|
pmiX |
x component of the
principal moment of inertia (external coordinates). |
|
pmiY |
y component of the
principal moment of inertia (external coordinates). |
|
pmiZ |
z component of the
principal moment of inertia (external coordinates). |
|
rgyr |
Radius of gyration. |
|
std_dim1 |
Standard dimension 1: the square root of the
largest eigenvalue of the covariance matrix of the
atomic coordinates. A standard dimension is equivalent to the standard
deviation along a principal component axis. |
|
std_dim2 |
Standard dimension 2: the square root of the second
largest eigenvalue of the covariance matrix of the
atomic coordinates. A standard dimension is equivalent to the standard
deviation along a principal component axis. |
|
std_dim3 |
Standard dimension 3: the square root of the
third largest eigenvalue of the covariance matrix
of the atomic coordinates. A standard dimension is equivalent to the standard
deviation along a principal component axis. |
|
vol |
van der Waals volume calculated using a grid approximation
(spacing 0.75 A). |
|
VSA |
van der Waals surface area. A polyhedral representation is used
for each atom in calculating the surface area. |
|
vsurf_V |
Interaction field volume |
|
vsurf_S |
Interaction field surface area |
|
vsurf_S |
Surface rugosity |
|
vsurf_S |
Surface globularity |
|
vsurf_W* |
Hydrophilic volume (8 descriptors) |
|
vsurf_IW* |
Hydrophilic integy moment (8 descriptors) |
|
vsurf_CW* |
Capacity factor (8 descriptors) |
|
vsurf_EWmin* |
Lowest hydrophilic energy (3 descriptors) |
|
vsurf_DW* |
Contact distances of vsurf_EWmin (3 descriptors) |
|
vsurf_D* |
Hydrophobic volume (8 descriptors) |
|
vsurf_ID* |
Hydrophobic integy moment (8 descriptors) |
|
vsurf_EDmin* |
Lowest hydrophobic energy (3 descriptors) |
|
vsurf_DD* |
Contact distances of vsurf_DDmin (3 descriptors) |
|
vsurf_HL* |
Hydrophilic-Lipophilic (2 descriptors) |
|
vsurf_A |
Amphiphilic moment |
|
vsurf_CA |
Critical packing parameter |
|
vsurf_Wp* |
Polar volume (8 descriptors) |
|
vsurf_HB1* |
H-bond donor capacity (8 descriptors) |
Conformation
Dependent Charge Descriptors
The following descriptors depend upon the stored partial charges of the molecules
and their conformations. Accessible surface area refers to the water accessible
surface (in Å2) area using a probe radius of 1.4 Å. Let qi denote the partial charge of atom i.
|
Code |
Description |
|
ASA+ |
Water accessible surface area of all atoms
with positive partial charge (strictly greater than 0). |
|
ASA- |
Water accessible surface area of all atoms
with negative partial charge (strictly less than 0). |
|
ASA_H |
Water accessible surface area of all
hydrophobic (|qi|<0.2)
atoms. |
|
ASA_P |
Water accessible surface area of all polar (|qi|>=0.2) atoms. |
|
DASA |
Absolute value of the difference between ASA+ and ASA-. |
|
CASA+ |
Positive charge weighted surface area, ASA+ times max { qi > 0 } [ |
|
CASA- |
Negative charge weighted surface area, ASA- times max { qi < 0 } [ |
|
DCASA |
Absolute value of the difference between CASA+ and CASA-
[Stanton 1990]. |
|
dipole |
Dipole moment calculated from the partial charges
of the molecule. |
|
dipoleX |
The x component of the dipole moment
(external coordinates). |
|
dipoleY |
The y component of the dipole moment
(external coordinates). |
|
dipoleZ |
The z component of the dipole moment
(external coordinates). |
|
FASA+ |
Fractional ASA+ calculated as ASA+ / ASA. |
|
FASA- |
Fractional ASA- calculated as ASA- / ASA. |
|
FCASA+ |
Fractional CASA+ calculated as CASA+ / ASA. |
|
FCASA- |
Fractional CASA- calculated as CASA- / ASA. |
|
FASA_H |
Fractional ASA_H calculated as ASA_H / ASA. |
|
FASA_P |
Fractional ASA_P calculated as ASA_P / ASA. |
References
|
[Balaban 1979] |
Balaban, A.T.; Five New Topological Indices for the Branching of Tree-Like
Graphs; Theoretica Chimica
Acta 53 (1979) 355–375. |
|
[Balaban 1982] |
Balaban, A.T.; Highly Discriminating Distance-Based Topological Index; Chemical
Physics Letters 89 No. 5 (1982) 399–404. |
|
[CRC 1994] |
CRC Handbook of Chemistry and Physics. CRC Press (1994). |
|
[Crippen 1999] |
|
|
[Cruciani 2000] |
Cruciani, G., Crivori, P., Carrupt, P.-A., Testa, B.; Molecular Fields in Quantitative Structure-Permeation Relationships: the VolSurf Approach; J. Mol. Struct. (Theochem) 503 (2000) 17–30. |
|
[Gasteiger 1980] |
Gasteiger, J., Marsili, M.; Iterative Partial
Equalization of Orbital Electronegativity - A Rapid
Access to Atomic Charges; Tetrahedron 36 (1980) 3219. |
|
[Ertl 2000] |
Ertl, P., Rohde, B., Selzer, P.; Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties; J. Med. Chem. 43 (2000) 3714–3717. |
|
[Hall 1991] |
Hall, L.H., Kier, L.B.; The Molecular
Connectivity Chi Indices and Kappa Shape Indices in Structure-Property Modeling; Reviews of Computational Chemistry 2
(1991). |
|
[Hall 1977] |
Hall, L.H., Kier, L.B.; The Nature of Structure-Activity Relationships and Their Relation to Molecular Connectivity; Eur. J. Med. Chem 12 (1977) 307. |
|
[Hou 2004] |
Hou, T.J., Xia, K., Zhang, W., Xu, X.J.; ADME Evaluation in Drug Discovery. 4. Prediction of Aqueous Solubility Based on Atom Contribution Approach; J. Chem. Inf. Comput. Sci. 44 (2004) 266–275. |
|
[Lipinski 1997] |
|
|
[LOGP 1998] |
Labute, P.; MOE LogP(Octanol/Water)
Model unpublished. Source code in $MOE/lib/svl/quasar.svl/q_logp.svl (1998). |
|
[MOPAC] |
Stewart, J.J.P.; MOPAC Manual (Seventh
Edition); 1993. |
|
[MREF 1998] |
Labute, P.; MOE Molar Refractivity Model unpublished. Source code in $MOE/lib/svl/quasar.svl/q_mref.svl (1998). |
|
[Oprea 2000] |
Oprea, |
|
[Pearlman 1998] |
Pearlman, R.S., Smith, K.M.; Novel Software Tools for Chemical Diversity; Persp. Drug. Disc. Des. 9/10/11 (1998) 339–353. |
|
[Petitjean 1992] |
Petitjean, M.; Applications of the Radius-Diameter Diagram to the Classification of Topological and Geometrical Shapes of Chemical Compounds; J. Chem. Inf. Comput. Sci. 32 (1992) 331–337. |
|
[Stanton 1990] |
Stanton, D., Jurs, P.; Development and Use of Charged Partial Surface-Area Structural Descriptors in Computer-Assisted Quantitative Structure-Property Relationship Studies; Anal. Chem. 62 (1990) 2323–2329. |
|
[Wiener 1947] |
Wiener, H.; Structural Determination of
Paraffin Boiling Points; Journal of the American Chemical Society 69
(1947) 17–20. |
![]() Copyright
© 1997-2008 Chemical Computing Group Inc.
Copyright
© 1997-2008 Chemical Computing Group Inc.
info@chemcomp.com